TRISOMÍA 21
El Síndrome de Down (SD), también llamado trisomía 21, es la causa más frecuente de retraso mental identificable de origen genético. Se trata de una anomalía cromosómica que tiene una incidencia de 1 de cada 800 nacidos, y que aumenta con la edad materna. Es la cromosomopatía más frecuente y mejor conocida.
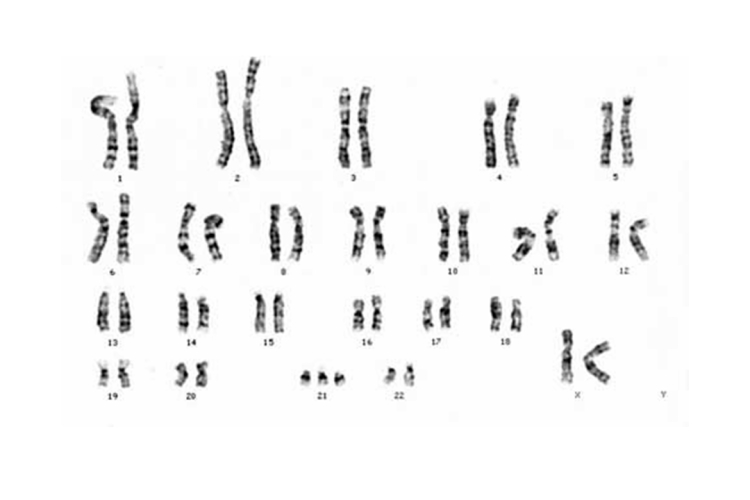
Fue descrito por John Langdon Down en 1866, dentro de su propuesta de clasificación de pacientes con discapacidad intelectual. Se asoció por primera vez con una alteración cromosómica en 1959, cuando Lejeune, Gautier y Turpin describieron 5 niños y 4 niñas con discapacidad intelectual y 47 cromosomas en el cultivo de fibroblastos, siendo un acrocéntrico pequeño el cromosoma extra. Los autores propusieron que el origen de este cromosoma extra se debía probablemente a una falta de disyunción, que por lo tanto ésta era la razón por la que la frecuencia del padecimiento aumentaba con la edad materna.
DIAGNÓSTICO
El diagnóstico es clínico y se confirma por citogenética. El patrón de características físicas observables (Gestalt) es altamente sugestivo, así como las alteraciones sistémicas. Sin embargo, no todas las alteraciones están presentes en cada individuo afectado. En recién nacidos el diagnóstico puede dificultarse; no obstante, diez características son altamente prevalentes. Hall, en 1966, analizó 48 recién nacidos afectados y encontró que 100% tuvieron 4 o más características y 89% tuvieron 6 o más.
a) Pruebas exploratorias o de cribado
Son de dos tipos:
Análisis ecográfico y análisis bioquí-micos de sangre.
Cribado prenatal ecográfico: Dentro de las técnicas de
cribado prenatal no invasivo la ecografía es uno de
los más importantes y extendidos. La ecografía se realiza
con un equipo que emite ondas de sonido de alta
frecuencia hacia las estructuras corporales, y recoge los
ecos provocados para proyectarlos como imágenes.
La prueba ecográfica se inicia muy tempranamente
en el embarazo (10-12 semanas). Son múltiples los
indicadores (marcadores) ecográficos que pueden
hacer sospechar que el feto puede tener síndrome de
Down, aunque no todos ellos tienen el mismo valor pronóstico. Uno de los marcadores más potente es el
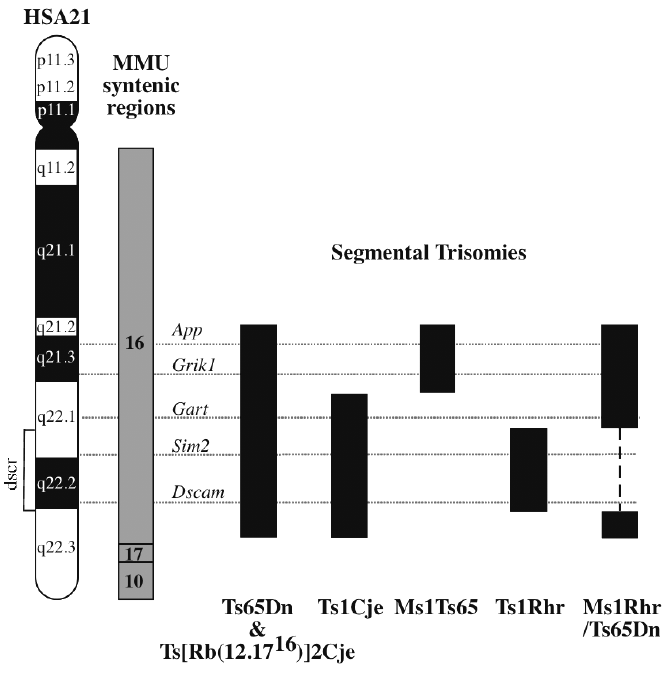
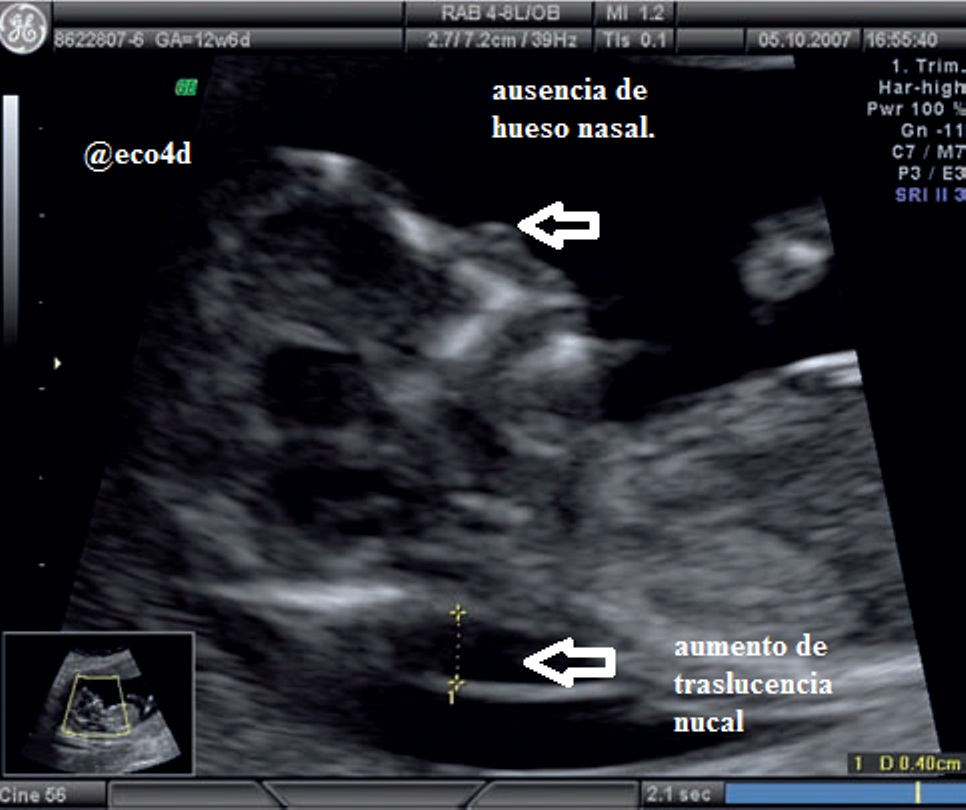
pliegue o traslucencia nucal que indica la presencia de
un pliegue en la nuca más grueso de lo normal, debido
a la acumulación de líquido subcutáneo en la nuca
del bebé. No es un signo definitivo de síndrome de
Down sino que puede aparecer con otros trastornos
cromosómicos. Se considera que un feto tiene un pliegue
engrosado cuando la medición de la distancia
entre la piel de la nuca y el hueso supera el percentil
95. Se han de realizar cálculos estadísticos que tengan
también en cuenta la edad de la madre y la semana
de gestación. Esta prueba ha de ser realizada por
ecografistas con experiencia, que posean un equipo
emisor y receptor de alta resolución y tecnología avanzada.
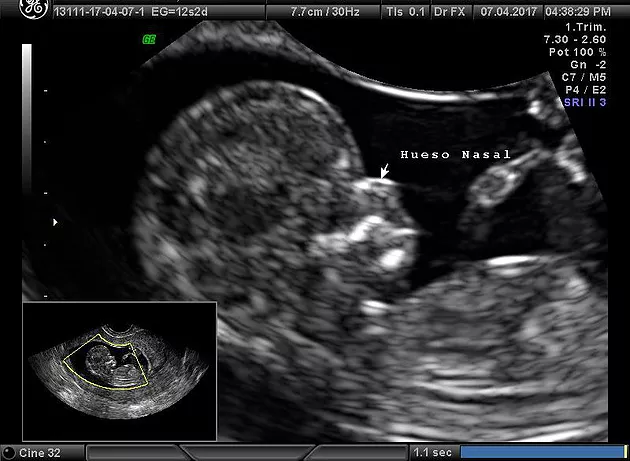
Otro de los parámetros que está adquiriendo
importancia es la presencia o no de osificación del
hueso de la nariz fetal; si el hueso no está formado a
las doce semanas es más probable que se trate de un
feto con síndrome de Down. Las cardiopatías congénitas son frecuentes en el síndrome de Down y muchas
de ellas son detectables in utero mediante ecocardiografía
fetal.
Cribado bioquímico: Se basa en la determinación en
suero materno de una serie de sustancias bioquímicas
de origen fetal o placentario. El cribado debe conjugarse
con la edad materna, ajustada al momento del parto,
y ser calibrado según la semana de gestación para
establecer la probabilidad de que el feto tenga síndrome
de Down.
El cribado puede ser de dos tipos, dependiendo de cuándo
se realice: en el primer trimestre de gestación, el
estudio más extendido es la valoración de los niveles
en suero materno de la PAPP-A (Pregnancy Associated
Placental Protein – A) y el nivel de la fracción b libre de
la HCG o free b (Hormona Gonadotropina Coriónica).
Este análisis se debe realizar entre las 8 y 12 semanas
de gestación (SG). Los valores se expresan en términos
absolutos, pero para su ponderación informática se han
de trasformar en valores relativos, los MoM (Multiples
of the Media) o valores de la mediana, establecidos
para cada semana de gestación ajustados según la ecografía.
Para aumentar su precisión, el cribado de primer
trimestre puede complementarse con los valores del
pliegue nucal medidos en MoM en la ecografía realizada
en la 12 semana de gestación, es el conocido como
cribado combinado, conjuntando así edad materna,
bioquímica y ecografía.
En el cribado de segundo trimestre, la extracción de
sangre materna ha de realizarse entre las 14 y 17 semana
de gestación ecográfica, de preferencia en la 15-16.
En este caso se sustituye la PAPP-A por los niveles de
a-fetoproteína, y en ocasiones además se valoran los
niveles de estriol. Los valores elevados de a-fetoproteína superiores a 3 MoM, pueden ser indicativos de que
el feto tenga un defecto del tubo neural, espina bífida.
El resultado del cribado es un coeficiente de riesgo,
una posibilidad sobre X de que ese feto tenga el síndrome
de Down. Se considera que un riesgo es alto
cuando es superior a 1/250; así una posibilidad entre
cien (1/100) sería un riesgo alto y una entre quinientas
sería un riesgo bajo. Se trata de un sistema de pruebas
de cribado o de selección poblacional, en ningún
caso son pruebas diagnósticas de síndrome de Down.
b) Pruebas diagnósticas
Requieren métodos invasivos cuya finalidad es la obtención
de una muestra de tejido fetal. Aunque son múltiples
los tejidos fetales que se pueden obtener, los que
por su accesibilidad son más fáciles de conseguir son
tres: la placenta o corion, el líquido amniótico y la sangre
fetal. Así tendremos respectivamente la biopsia de
corion (BC), la amniocentesis y la cordocentesis.
Biopsia de corion. La BC consiste en la obtención de una
muestra de corion, que es el tejido que posteriormente
constituirá la placenta. Este tejido ha de tener la misma
información genética que el feto, por proceder ambos de
la misma célula original. Según la vía de acceso al tejido
corial, el riesgo de pérdida fetal atribuible a este sistema
de muestreo es de un 1% cuando lo practican
profesionales experimentados. Este riesgo debe añadirse
al de pérdida fetal propio del embarazo durante un
periodo de unos días. La BC tiene que realizarse preferentemente
entre las semanas 10 y 13 de gestación.
Los resultados se obtienen al cabo de unos pocos días,
entre 2-7 días para el análisis directo de la muestra y 12-
15 días para los cultivados.
Amniocentesis. Consiste en la obtención de una muestra
de líquido amniótico, en el que hay células de descamación
fetal, por lo tanto con la misma dotación cromosómica
que el feto. La vía de acceso al líquido es
por punción abdominal, siempre con control ecográfico.
Esta técnica tiene un riesgo de pérdida fetal situado
entre un 0,5% y un 1% cuando es realizada por profesionales
debidamente entrenados; este riesgo incrementa
durante unos días el riesgo de perdida fetal propio
del embarazo. La amniocentesis debe de realizarse
preferentemente después de la semana 15 de gestación,
ya que la poca cantidad de líquido amniótico existente
antes de esa semana y el consiguiente incremento
de riesgo de pérdida fetal desaconsejan realizar la
prueba en periodos más precoces. Al igual que en la
BC, se trata de un análisis fetal indirecto, pues se estudian
células que proceden de la célula original, por lo tanto
si la primera tenía una trisomía 21 u otra alteración
cromosómica, se verá reflejada en la muestra. La fiabilidad
es muy alta, superior al 99%. Las células de la
muestra pueden analizarse directamente en 24-48
horas, por los métodos de FISH o PCR. El resultado
definitivo es el estudio citogenético (cariotipo), para lo
cual es preciso un cultivo celular, en cuyo caso el análisis puede demorarse entre 12-18 días.
Cordocentesis. Es un método extraordinario que solo
se utiliza en casos excepcionales. Consiste en la punción
del cordón umbilical a través de la pared abdominal de
la madre, para la obtención de sangre fetal; en el caso
de síndrome de Down sería para el estudio del cariotipo
fetal. En comparación con los otros métodos, su riesgo
de pérdida fetal es alto, situado en el 3% cuando la
punción se realiza en un centro experimentado. Esta
prueba no se recomienda hacerla antes de las 20 semanas
de gestación. Los resultados cromosómicos suelen
tenerse antes de una semana.
Análisis de ADN fetal en la sangre de la madre. Es una
técnica en fase experimental que permitirá un diagnóstico
preciso y relativamente precoz por un método no
invasivo. Requiere alta tecnología.